1/1
¥200假一赔十
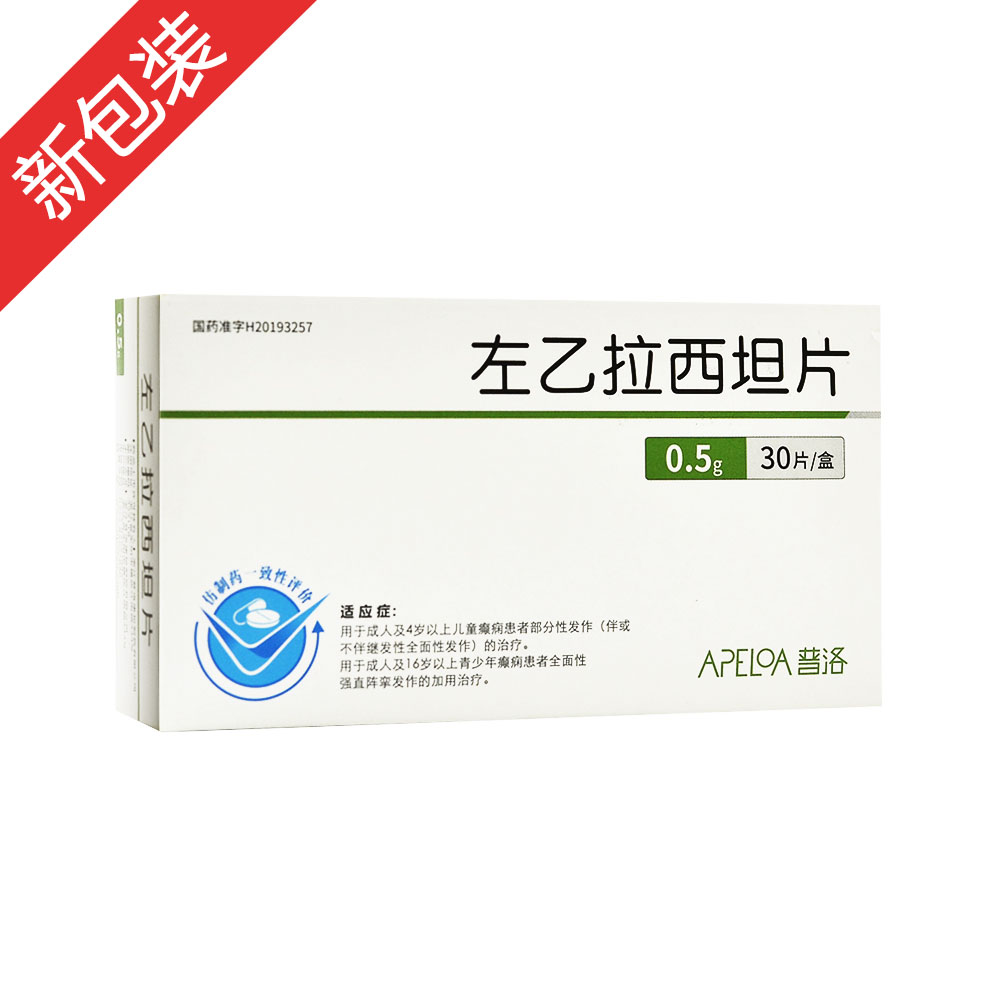
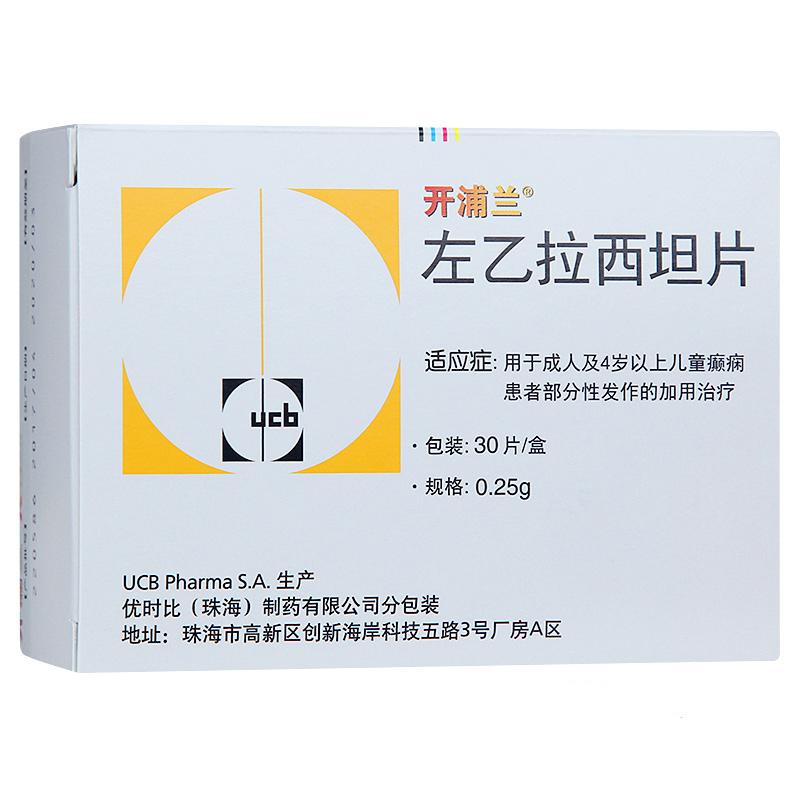
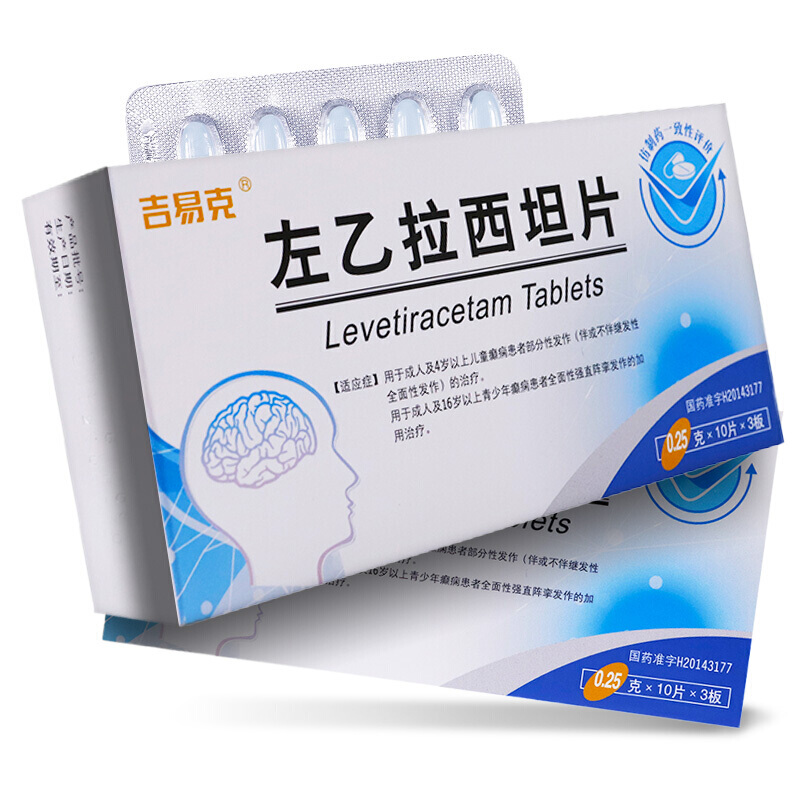
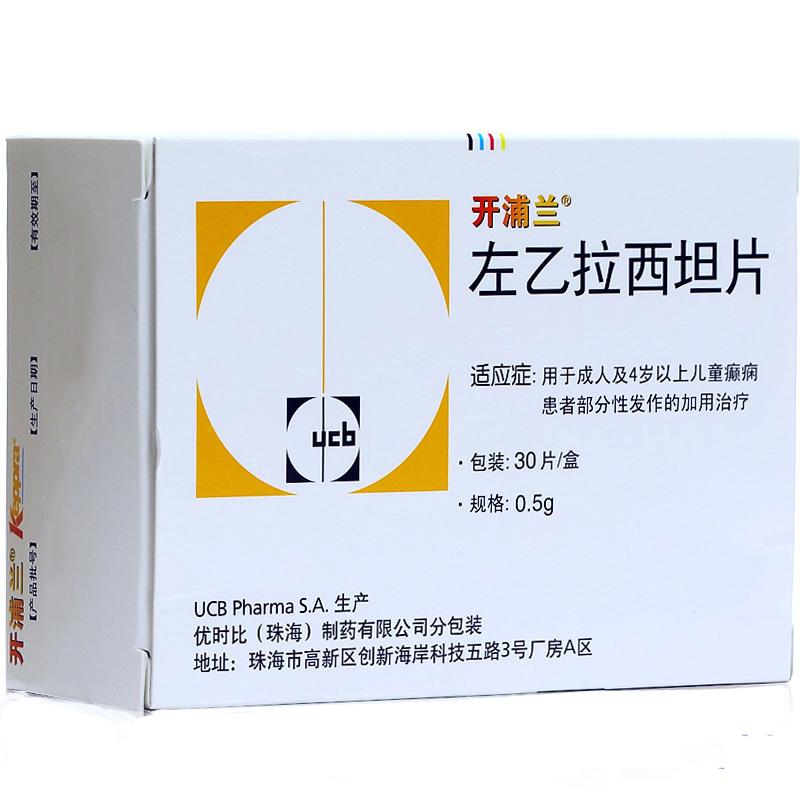
左乙拉西坦片(开浦兰)
处方药 | 须凭处方在药师指导下购买和使用
批号注册证号H20160251
规格0.5g*30s
厂家ucb pharma s.a.(比利时)

| 产品品名 | 左乙拉西坦片(开浦兰) |
| 主要原料 | 左乙拉西坦。 |
| 主要作用 | 用于成人及4岁以上儿童癫痫患者部分性发作的加用治疗。 |
| 产品规格 | 0.5g*30s |
| 用法用量 | (1)给药途径 :口服。需以适量的水吞服,服用不受进食影响。(2)给药方法和剂量 :成人(>18岁)和青少年(12-17岁)体重≥50kg 起始治疗剂量为每次500mg,每日2次。根据临床效果及耐受性,每日剂量可增加至每次1500mg,每日2次。其他请详见说明书。 |
| 生产企业 | ucb pharma s.a.(比利时) |